
Introduction to QSAR Bioactivity Predictor
QSAR Bioactivity Predictor is a machine learning-based tool that models the quantitative relationship between chemical structure and biological activity to predict compound bioactivity. This approach is fundamental in modern drug discovery for lead compound screening, target validation, and drug repurposing.
The tool employs a sophisticated algorithm that begins by calculating up to 1800 2D/3D molecular descriptors using RDKit. It then utilizes advanced feature selection methods and a suite of machine learning models—including Random Forest, XGBoost, and Stacking Ensembles—to build a robust predictive model. The final output provides predicted bioactivity values (e.g., IC₅₀, EC₅₀), along with comprehensive model performance metrics to support rational, data-driven decision-making in compound optimization.
A Step-by-Step Guide to Execute the Tool
Welcome to the QSAR Bioactivity Predictor workspace. Before you begin, you must create a JOB ID by clicking the "Create Job ID" button.
TIP: You will not be able to access any tool features until a JOB ID has been created.
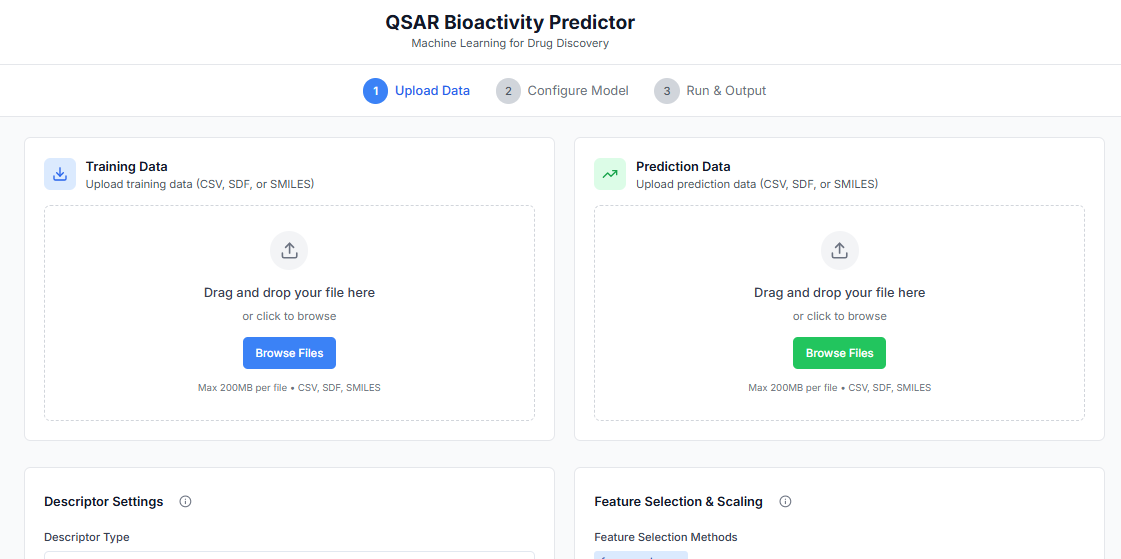
First, upload the training data. This file must be target-specific and contain columns for SMILES strings and their corresponding biological activity values, along with the SI unit.
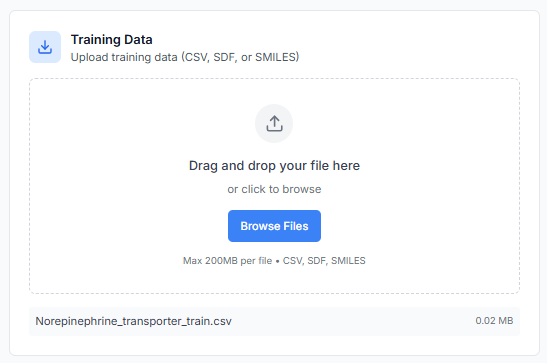
Along with the training set, you must also upload the prediction data. This file should contain the SMILES strings of the new, unlabeled compounds you wish to predict the activity for.
Configuring the QSAR Model
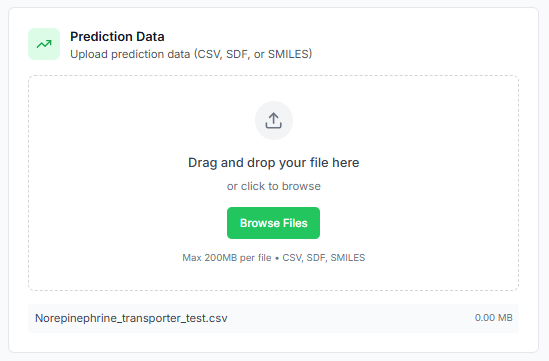
Next, define the number of molecular descriptors to be generated for each molecule. You can also specify the type of descriptors to use, choosing between 2D, 3D, or both.
In this step, select the feature selection method you want to apply. Multiple methods can be chosen. You also need to select a scaling method to normalize the descriptor data before training.
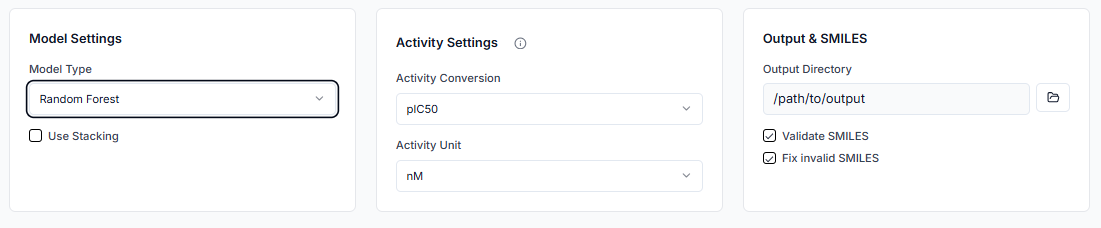
Now, select the ML model to be used for training. If you want to use a stacking ensemble, you can select multiple ML models. You must also specify the SI unit of the biological activity (e.g., IC50, pIC50) and its unit (e.g., nM, µM). Finally, indicate whether to validate SMILES before processing.
Tip: Keeping the SMILES validation option checked is recommended to ensure data quality and prevent errors during model training.
Running the Prediction and Evaluating the Model
With all the data uploaded and parameters configured, you are ready to build the model. Click on 'Run Prediction' to start the process.

Once the process is complete, the results will be displayed. First, look into the training and prediction dataset statistics to get an overview of the data used in your model.

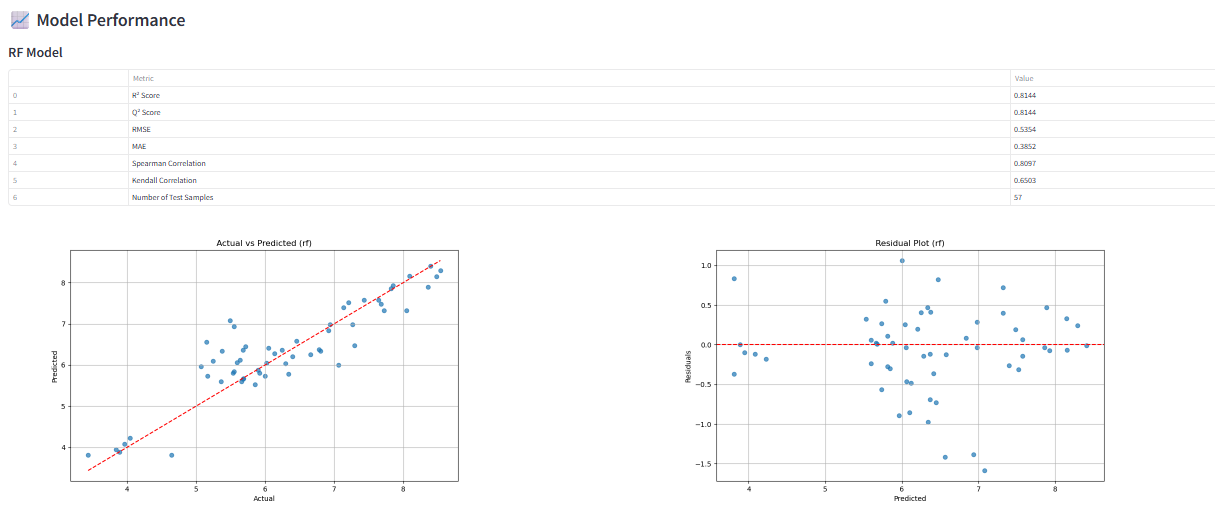
Next, evaluate the model performance. The tool provides a detailed report with key evaluation metrics (e.g., R², Q², RMSE) and plots, allowing you to assess the accuracy and predictive power of your QSAR model.
Analyzing the Final Predictions
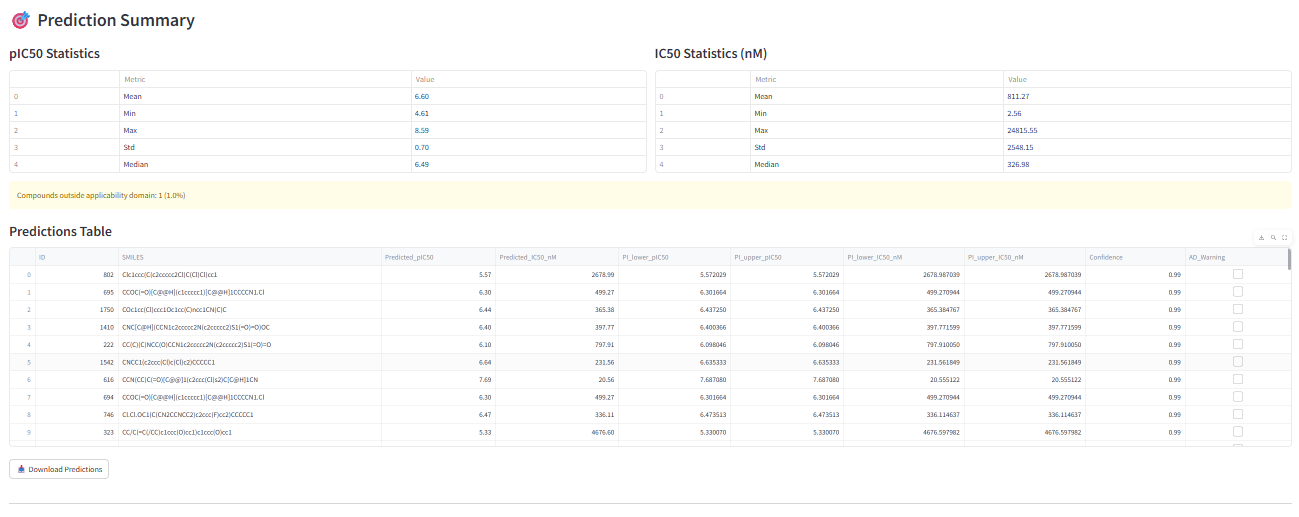
Finally, you can analyze the predictions for your new compounds. The results table will show each molecule from your prediction set along with its predicted bioactivity value.
So, by using the QSAR Bioactivity Predictor, you have successfully built, evaluated, and used a custom QSAR model to predict the biological activity of new chemical compounds.