
Introduction to FastQC
FastQC is a fundamental bioinformatics tool for quality control of high-throughput sequencing data. It rapidly analyzes raw sequence files to generate clear graphical reports on key metrics like base quality, GC content and adapter contamination. These reports allow researchers to quickly diagnose the health of their sequencing data before analysis.
By identifying potential issues such as low-quality reads or technical artifacts, FastQC ensures data integrity, guiding necessary pre-processing steps and improving the reliability of downstream applications like variant calling and RNA-seq. This makes it an essential first step in virtually any genomics workflow.
A Step-by-Step Guide to Execute the Tool
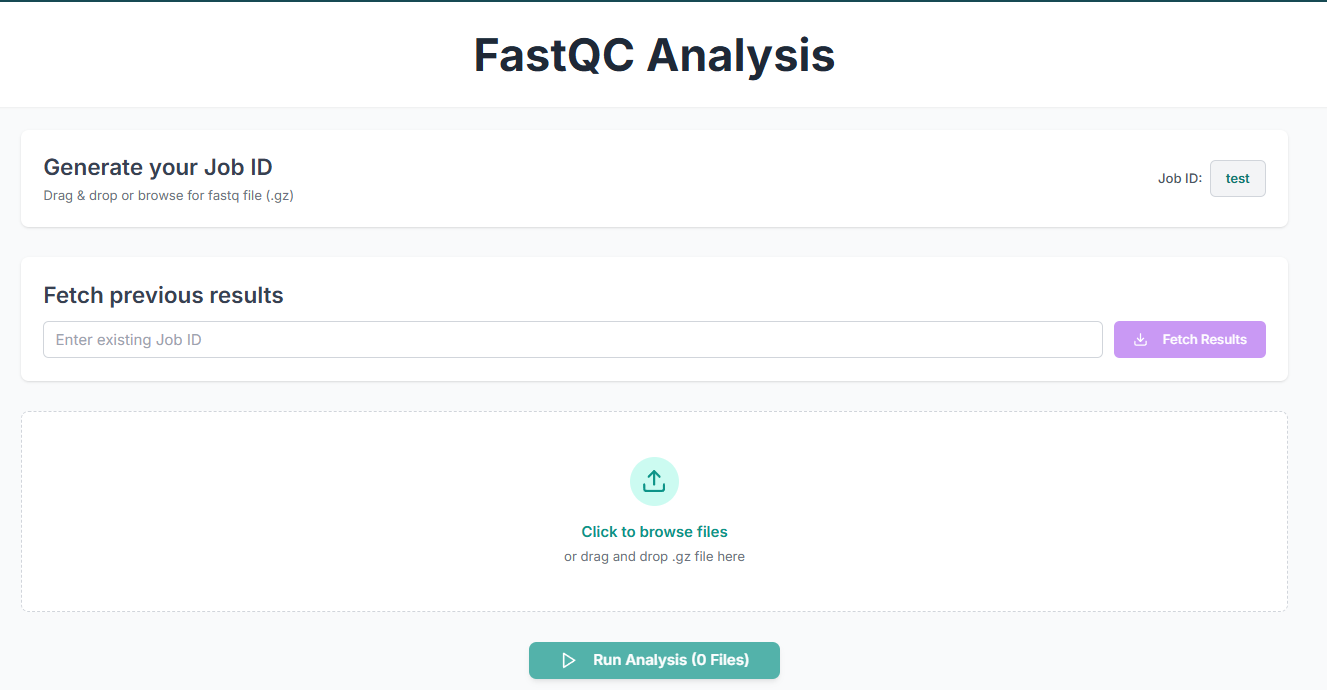
Before executing the tool, you must create a Job ID. You can customize this ID, or the job ID will be created by the tool on its own.
TIP: Without creating a JOB ID, you will not be able to access any options of the tool.

Alternatively, you can fetch previous results by entering an existing job ID.
This is the FastQC tool's application workspace page, where various options for analysing sequences are available. This tutorial will explain each option in detail.
Uploading Files and Running the Analysis
Upload the fastq files in .fastq or .fastq.gz format by clicking on "Click to browse files".
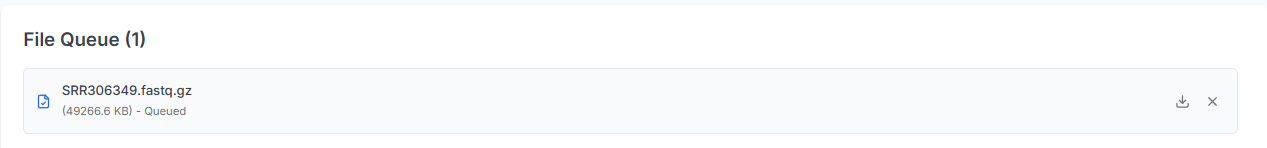
The uploaded files and the number of files will be displayed below.
Click on "Run Analysis".
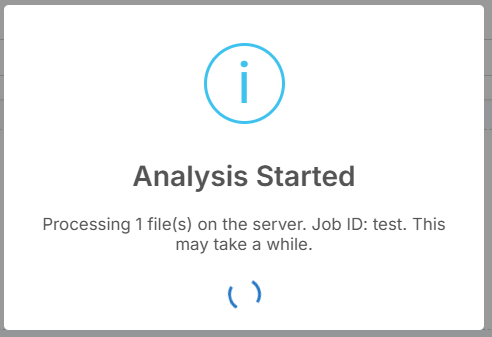
After clicking "Run Analysis", a pop-up will appear indicating that the analysis has started.
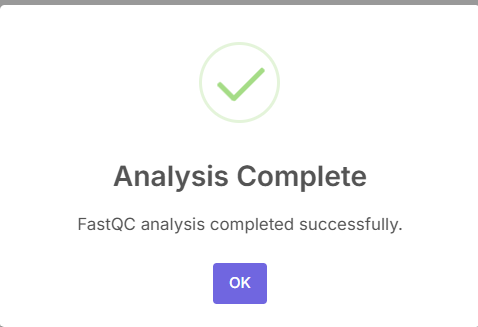
After the analysis has been completed, another pop-up will appear.
Analyzing the QC Report
The plots after analysis will be shown below.
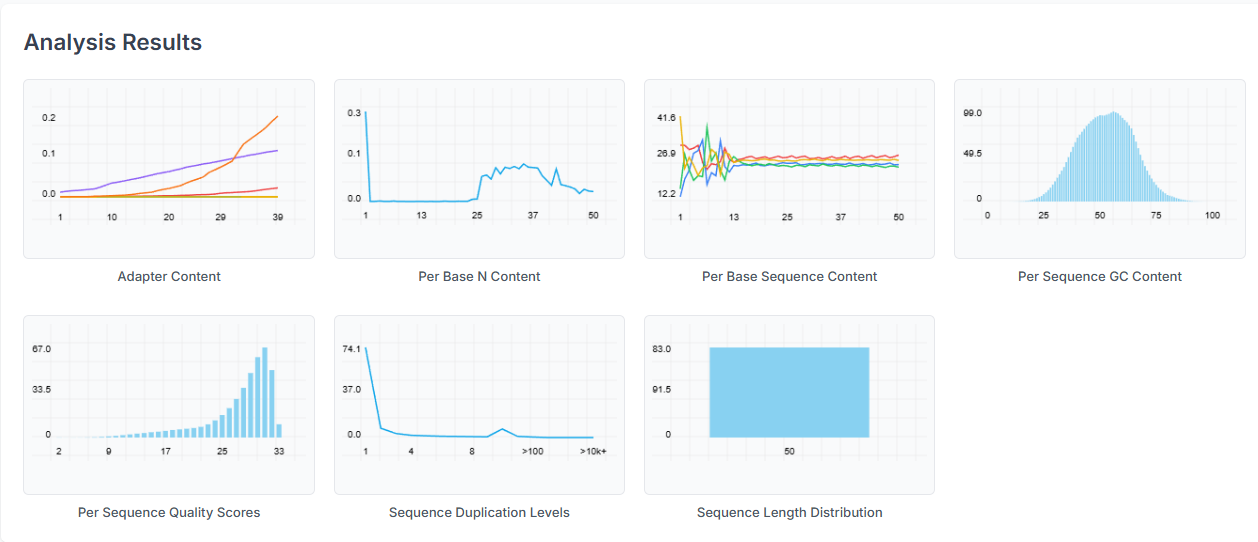
The Summary table provides an overview of the status for each parameter, indicating whether it has passed, failed, or triggered a warning.
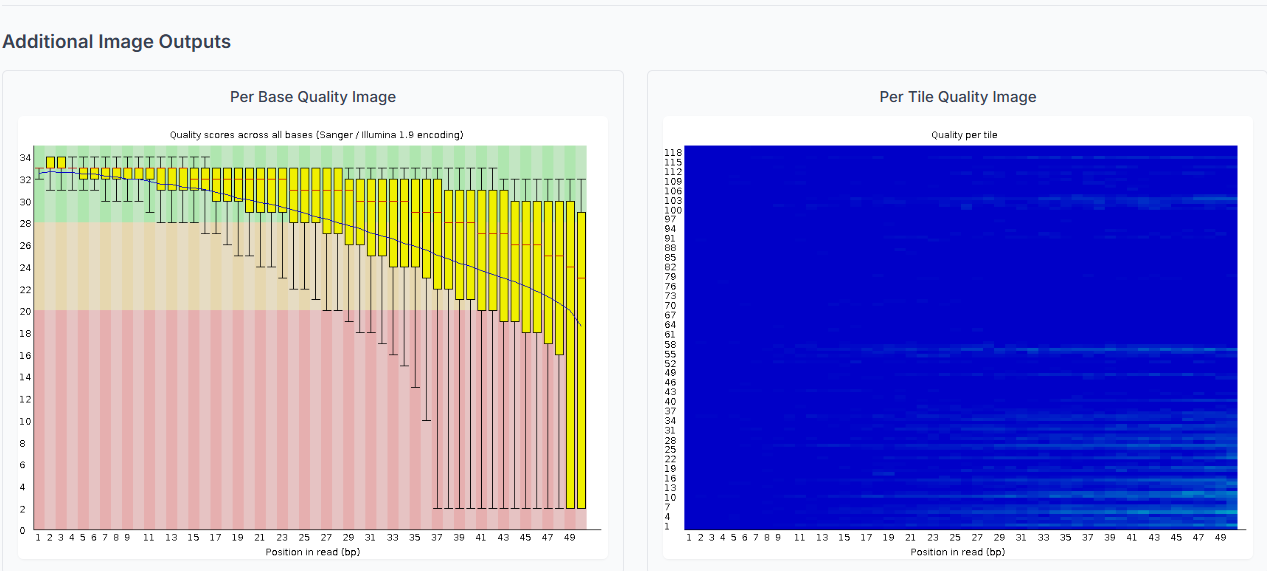
FastQC has successfully completed its execution with a vital assessment of your raw sequencing data. Understanding each parameter in the report is important. This early identification of potential issues safeguards that you can make informed decisions on data trimming and filtering for accurate results for all subsequent bioinformatics analyses.