
Introduction to Molecular Dynamics Simulation (MD)
Molecular Dynamics (MD) is a powerful computational technique that simulates the physical movements of atoms and molecules over time by numerically solving Newton's laws of motion. Governed by a set of parameters known as a force field, the simulation calculates the interactions between particles to generate a trajectory, or "molecular movie," that reveals how a system evolves.
Key algorithms like the Verlet integrator, Particle Mesh Ewald for electrostatics and various thermostats and barostats are used to ensure physical realism and control environmental conditions. MD is widely applied to study complex biological phenomena, including protein folding, ligand binding with its primary outputs being trajectory, energy and log files that allow for detailed analysis of conformational changes, system stability and thermodynamic properties.
A Step-by-Step Guide to Execute the Tool
1. Choose Your Simulation Type
- Protein-Ligand Simulation: Used when you're analyzing the interaction, stability or dynamics of a ligand (small molecule) bound to a protein receptor.
- Only Protein Simulation: Used for simulating the dynamics, folding or stability of a standalone protein.
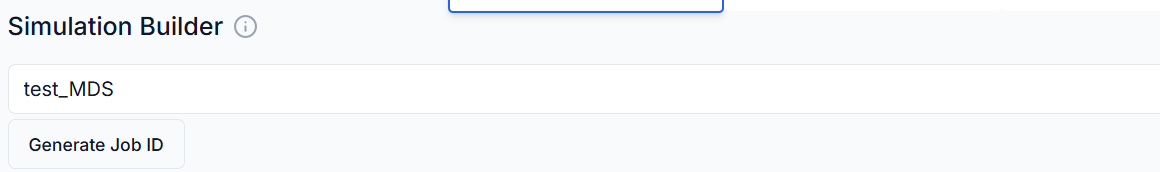
2. Create or Enter Job ID
- New Job: Leave the job ID field blank to generate a new unique job ID automatically.
- Existing Job: If resuming or continuing a previous simulation, enter the existing job ID.
Upon creation, a success message will appear.

3. Configure Simulation Parameters – Edit MDP Files
Five GROMACS compatible .mdp (Molecular Dynamics Parameters) files control different simulation phases:
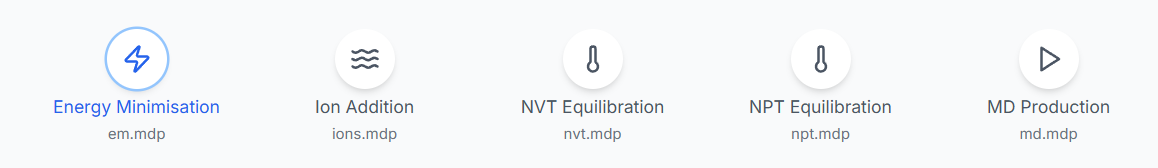
- Energy Minimization (em.mdp): Removes steric clashes, optimizes geometry.
- Ion Addition (ions.mdp): Prepares for system neutralization.
- NVT Equilibration (nvt.mdp): Maintains constant volume and temperature.
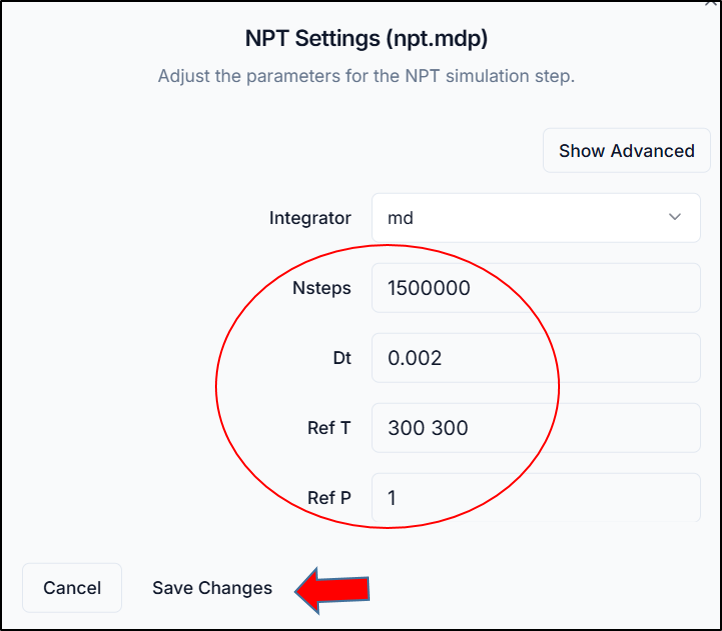
- NPT Equilibration (npt.mdp): Maintains constant pressure and temperature.
- Production Run (md.mdp): Full simulation phase to capture dynamics.
Click each .mdp file from the list, make necessary parameter adjustments (e.g., temperature, timestep, duration), and click "Save Changes" for each file.
4. Choose the Force Field
The force field defines the atomic parameters and interaction rules used during simulation. From the Force Field drop-down, select the one compatible with your system and ligand parameters.

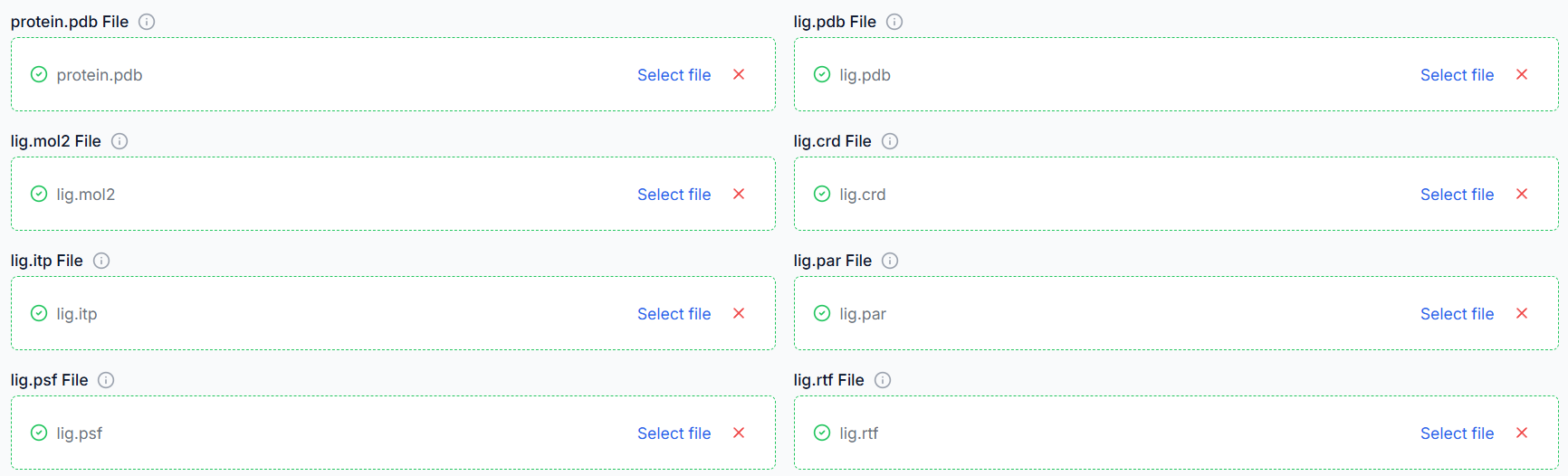
5. Upload Ligand Parameters (if protein-ligand simulation)
For protein-ligand simulations, topology and parameter files for the ligand (e.g., .itp, .prm, .top, .gro) are needed. Upload all ligand parameter files and ensure file names match and parameters align with the chosen force field.
Tip: Use SwissParam or Antechamber to generate ligand files compatible with GROMACS.
6. Upload Protein (if protein simulation)
For protein-only simulations, a protein file in .pdb format is needed.
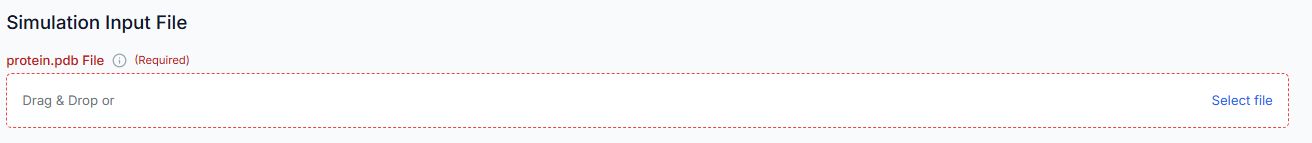
7. Submit Simulation
Once everything is configured (Parameters edited, Ligand files uploaded, Force field selected), click on "Submit Simulation" to start the backend processing and run the GROMACS simulation workflow.

8. Analysis
Once your simulation has successfully completed, the next step is to analyze the generated trajectory. To begin the analysis, you must first load the results from your simulation.

- Protein-Ligand Analysis: Used to investigate interaction dynamics, such as the stability of the ligand in the binding pocket and the conformational changes of the protein.
- Only Protein Analysis: Used to evaluate the intrinsic properties of the protein, such as its structural stability and flexibility over time.
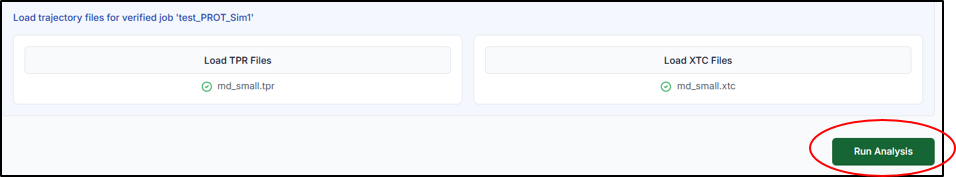
The system will automatically fetch the necessary output files, primarily the run-input file (.tpr) which contains system topology, and the compressed trajectory file (.xtc) which stores the atomic coordinates over time. Click on "Run Analysis" to start the calculations.
By following this tutorial, you have successfully navigated the complete workflow of a molecular dynamics simulation, from the initial setup and submission to the crucial post-simulation analysis.